Ever wondered how going off carbs for a couple of days almost always shows instant results? Thank all the glycogen stored in your body. But some people have trouble storing this entity as we do, disrupting their chance to live a typical life. Let’s peak into what actually happens with Glycogen Storage Disease.
Metabolism is a biological process that breaks down the food we eat and provides energy to keep us alive.
In some cases, different key stakeholders in this process fail to fulfill their duty.
There is a spectrum of conditions that fall under the umbrella of Glycogen Storage Disease (GSD), which cause such trouble.
Since it did not have any popular awareness challenge go viral (bring back the ice buckets!) and fortunately is a rare condition, it hasn’t had too much light thrown on it.
According to the reports, the incidence rates appear to be 1 in 100,000.
As the symptoms set out at an early age, Glycogen Storage Disease appears to affect the little ones more than the adults.
It’s complicated: Sugars

Our body uses glycogen, a complex sugar compound, as a fundamental storing unit of energy. Metabolizing glycogen, to break it down into glucose, provides the instant energy we need. As we need to conserve some of this energy, these glucose molecules are combined back into glycogen. This is used as a reservoir to tap into when there is a lack of energy supply. Different parts of the muscles and the liver act as storage units for glycogen.
Bad, bad enzymes, no donut for you
For this process to take place, some special proteins called enzymes (biocatalysts) aid the formation and deformation of glycogen.
When these enzymes don't function optimally, it leads to a spectrum of diseases.
This could lead to a range of different symptoms depending on the type of diseases.
The types are classified based on the enzyme which is at fault. It starts from GSD1 and runs up until GSD 15.
The types 1 to 4 cover almost 90% of the reported cases, with sub-type of GSD1 - GSD 1a, aka Von Gierke Disease being most common.
As the condition affects the necessity of food metabolism, symptoms start to show 3 to 4 months after birth.
What causes Glycogen Storage Diseases?
Get ready for some science-ing.
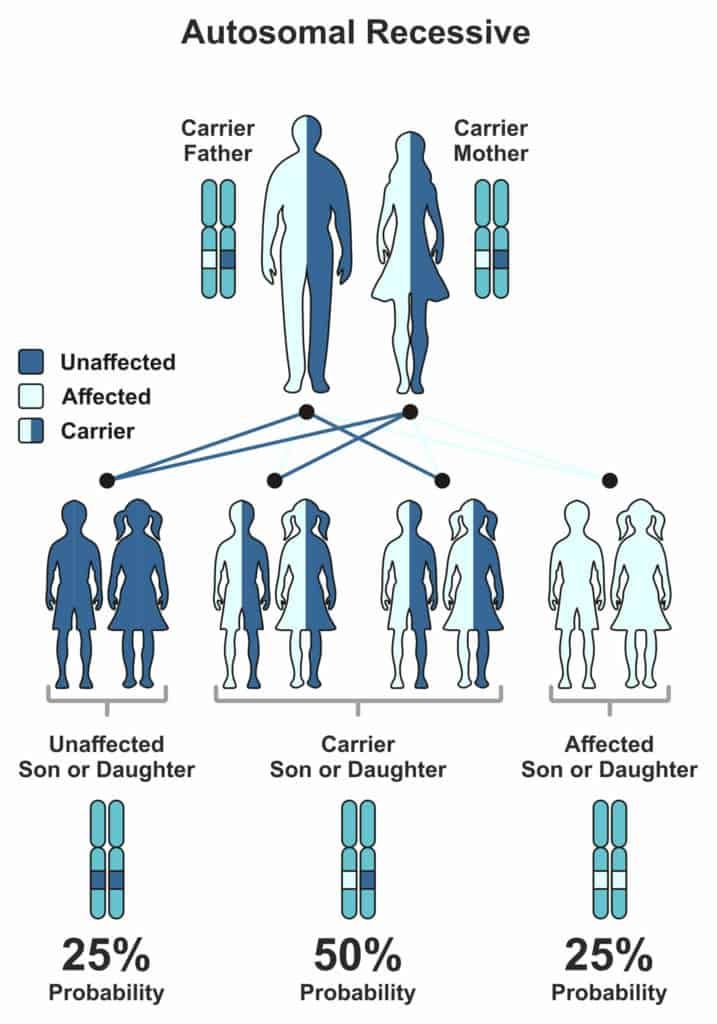
The cause of Glycogen Storage Disease is genetic.
The gene that is responsible for the malfunction of the enzyme can pass on from the parent generation to the next, making it hereditary.
But the child will express symptoms of the associated GSD, only when both parents possess a defective gene.
Every human gets their 23 chromosomes from each parent.
Genes that are subunits to chromosomes have one trait, which is dominant and other recessive (some exceptions, of course).
The level of expression marks the difference between them.
Now, for the expression of a recessive trait, both parents must pass on their recessive versions of traits.
The Mendelian world calls this an autosomal recessive condition.
A random process selects the gene to be passed down to the next generation.
In the case of autosomal recessive conditions, there is a 25% chance of occurrence.
If just one parent passes it on, then the child will remain healthy but acts as a carrier of the gene.
Carriers could potentially pass it on to the coming generations.
The journey of glycogen
Making of Glycogen
We obtain glucose from the diet we eat.
Glycogenin is an enzyme that is responsible for cutting down this glucose into short fragments.
Another enzyme, glycogen synthase, helps in the conversion of glucose into Glycogen.
Now, some branching enzymes add branches to Glycogen, which the liver then stores as a reservoir of energy.
Breaking of Glycogen
When we fast or there is a need for muscle contraction, the body taps into the said reservoir.
The process of breakdown involves four enzymes.
Glycogen Phosphorylase and glycogen debranching enzymes help in unraveling the molecule to release glucose and expend energy.
Another method of breaking it down involves enzymes such as a-glucosidase and Glucose-6-phosphatase.
Why do these enzymes misbehave?
It’s the teacher's fault.
Every action in our body is instructed by the beautifully wound helix, DNA.
As these enzymes correspond to specific genes, any defect in this gene will directly translate into the enzyme’s action.
Take the example of the most common type of GSD - GSD type 1a.
One of the enzyme genes involved in breaking down glycogen is Glucose-6-phosphatase. G6PC and SLC37A4 genes code for this enzyme.
As genes code for proteins and proteins like enzymes carry out the function, an error in the gene structure or function will lead to a collapse of the entire system.
A good analogy would be a loose brick in a building that could make it fall into pieces. Geneticists call these errors, mutations. As these ‘errors’ have been major contributors in the history of evolution, they have been beneficial in many ways.
Unfortunately, not in the case of Glycogen Storage Diseases.
So, when there is a mutation in this gene, glucose-6-phosphatase does not play its role and leads to the build-up of Glycogen and fat.
We know, too much of anything is toxic. Hence, the accumulation of Glycogen and fat hinders the function of organs like liver and kidney.
In the case of type 3 Cori disease/Forbes disease, a distinct part of the gene, called Exon3, carries two mutations that cause the debranching enzyme to malfunction.
As a debranching enzyme is responsible for the decomposition of Glycogen, it leads to toxic accumulation.
Andersen disease (type 4) affects the GBE1 gene that codes for glycogen branching enzyme leading to large amounts of abnormal Glycogen accumulated, causing severe conditions like liver cirrhosis, which is ‘doctor’ for scarring.
Types 1, 3, and 4 are far more common in comparison to other types.
What are the symptoms?
Most often, all different types have a combination of some common symptoms in varying intensities.
- As glucose is a key player, blood glucose levels drop (hypoglycemia).
- Liver enlargement leading to a swollen belly.
- Growth is affected, leading to short stature and thin bones, contributing to overall body weakness and muscle cramps.
Diagnosis of Glycogen Storage Diseases
As the age of onset is rather young in the case of Glycogen Storage Diseases, doctors generally ask the parents about symptoms showcased by their child and call for relevant tests.
Blood tests and MRI/ultrasound scans are routine. In some cases, a biopsy of a suspected organ might be required for confirmatory diagnosis.
Genetic testing has evolved over time and is performed to confirm the diagnosis.
This is extremely helpful in the case of couples who have observed a family history of Glycogen Storage Disease.
Seeking genetic counseling before planning a family will help provide a clearer picture of the chance of occurrence in their case.
Carrier testing for at-risk family members and prenatal diagnosis have led to significant changes in family planning.
Complex carbs is the solution
The treatment options are specific to the type of GSD diagnosed with the patient.
Generally, it includes major dietary restrictions.
Uncooked corn starch can be a good feed to children over two years of age, as corn starch can promise a slow release of glucose.
This is fed in small portions throughout the day.
For type 1, elimination of food which is high in lactose and fructose is advised (that's pretty much everything tasty, from mozzarella to maple syrup).
Allopurinol is prescribed if there is a risk of kidney stones or gout, as it reduces the levels of uric acid in the blood.
Some extreme cases like the type 4 of GSD could require liver transplant depending on the extent of the condition.
When the patient is prone to frequent muscle cramps during exercise, a high protein diet is advised.
In some cases, the intake of glucose and fructose is advised. And of course, as their muscles are now more susceptible to damage, over-exhausting the body is a big No-No.
Prevention and management
Unfortunately, prevention is barely an option.
As this is a genetic condition, the only way of prevention is to ‘not pass on the genes’.
Parents can seek genetic counseling to check if both carry the defective gene as it takes two to tango in this case.
But we have come a long way with the prognosis of the condition.
The unawareness of the disease’s details led medical researchers to believe GSD to be a fatal condition until the early ’70s.
People now live long and normal lives with significant dietary shifts.
This barely seems like a change in the millennial world of Keto and Atkins!
Speaking of Keto and Atkins, Glycogen is the secret quarterback in the low-carb diet game.
What is the effect of glycogen storage in healthy people?
Glycogen storage and weight loss
Glycogen storage and weight loss have a strange love story.
A typical healthy liver in an individual can hold up to 400 grams of glycogen and muscle cells, about 100 grams.
As glycogen and water co-exist in a 1:3 ratio, there are 3 grams of water present for every gram of glycogen.
This is the real reason behind the initial weight loss observed when any sort of low-carb diet is practiced.
It would rather be ideal for comparing body fat percentage or keeping track of monthly weight changes, for getting a better measure of body change as opposed to this initial quick loss of weight.
The body stores about 1500-2000 calories of Glycogen typically.
With a low-carb diet, the body uses this up and has little or no reservoir of energy. Possibly dehydrated.
This can lead to a constant state of fatigue in some extreme cases that could potentially damage the liver as it would be exposed to undue stress.
So we should be cautious when we make such lifestyle changes and always seek professional guidance.
In the case of sportsmen, a low-carb diet could lead to quick use of the stored Glycogen, especially the ones stored in the muscles.
This leads to a case of muscle fatigue and, in extreme cases, will lead to a phenomenon known as “hitting the wall”. Glycogen is also brain food.
So severe lack of Glycogen leads to cognitive symptoms like confusion, disorientation when you have a “bonk” during exercise.
What is the silver lining?
Though there is cutting current edge research to discover new enzyme replacement therapies and gene therapies, this spectrum of diseases affects many lives.
But once again, we are hopeful that genomics will save the day!
Have your raw data from 23andMe, FTDNA, or MyHeritage?
Upload your DNA raw data to Xcode Life for insights into 700+ health-related traits
References
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4146814/ - Ozen H. (2007). Glycogen storage diseases: new perspectives. World journal of gastroenterology, 13(18), 2541–2553. https://doi.org/10.3748/wjg.v13.i18.2541
https://www.tandfonline.com/doi/full/10.3109/01913123.2011.601404 - Hicks J, Wartchow E, Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011;35(5):183‐196. doi:10.3109/01913123.2011.601404
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(04)16986-9/fulltext Astrup, A., Meinert Larsen, T., & Harper, A. (2004). Atkins and other low-carbohydrate diets: hoax or an effective tool for weight loss?. Lancet (London, England), 364(9437), 897–899. https://doi.org/10.1016/S0140-6736(04)16986-9
https://onlinelibrary.wiley.com/doi/abs/10.1038/icb.2015.109 - Gleeson M. (2016). Immunological aspects of sport nutrition. Immunology and cell biology, 94(2), 117–123. https://doi.org/10.1038/icb.2015.109


